Thank you for your visiting | Dr. Daisuke Aoki

研究内容 / Research
Research based on organic chemistry, polymer chemistry, and supramolecular chemistry
有機化学、高分子化学、超分子化学をベースとした研究を進めています
The Living Radical Polymerization Initiated from Polymerization Inhibitor
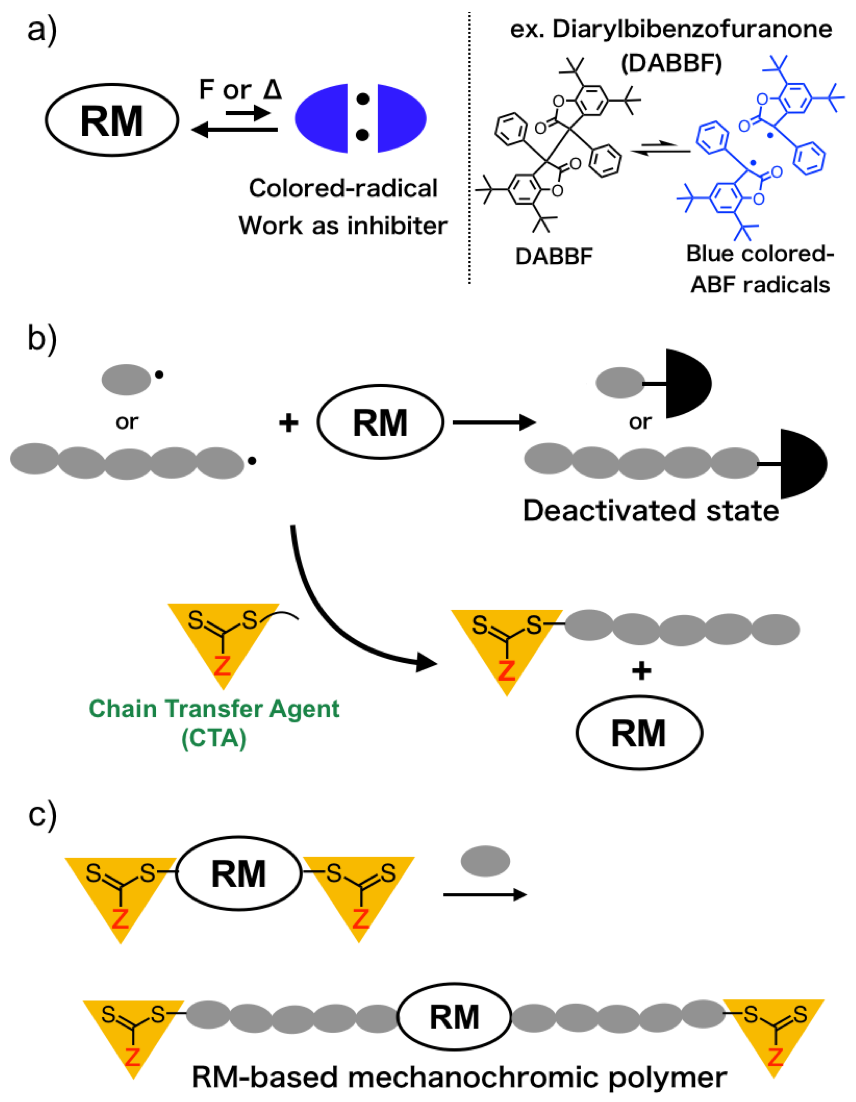
Radical-type mechanochromophores (RMs), which generate relatively stable and colored radical species upon mechanical stimulate, provide mechanochromic polymers with a quantitative evaluation of mechanochromic property by using the electron paramagnetic resonance (EPR) spectroscopy. However, the range of application of RMs is still limited by their equilibriums between the corresponding radicals that work as polymerization inhibitor, and RM cannot be used in combination with radical polymerization which enables wide range of vinyl monomers available. In this work, mechanochromic polymer based on RM was synthesized by using RAFT polymerization. The RAFT polymerization of vinyl monomers in the presence of a diarylbibenzofuranone (DABBF)-based RAFT agent which consists of a DABBF units used as a typical RM and two RAFT agents, successfully proceeded with a living manner to give the well-defined DABBF-centered polymers. The resulting polymer exhibited change in color based on the cleavage of central carbon-carbon bond of DABBF moiety, which is supported by solid UV-Vis absorption measurements and EPR measurements.
Plastics to Fertilizers: Chemical Recycling of a Bio-based Polycarbonate as a Fertilizer Source
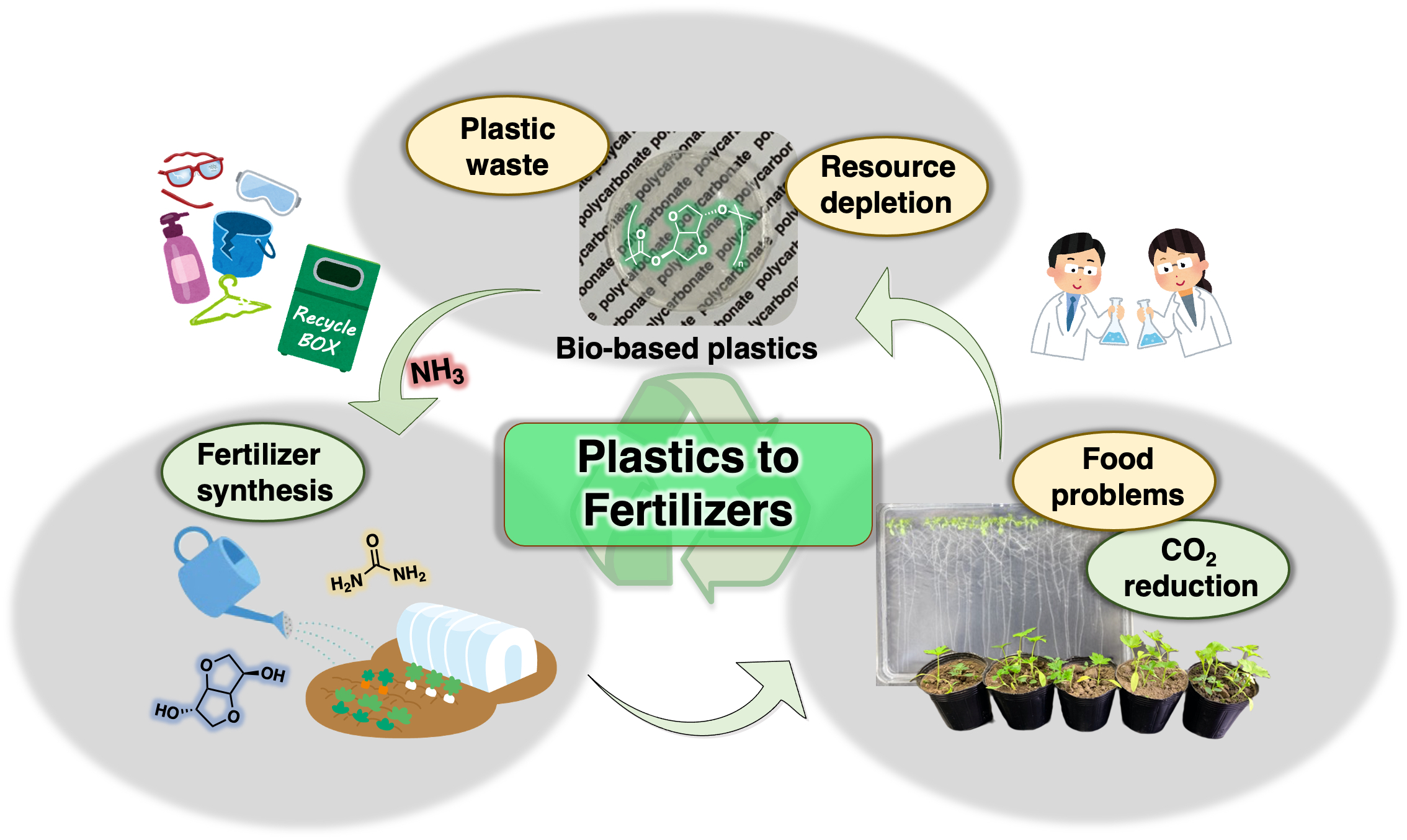
Commodity polymer materials are now required to be environmentally friendly due to problems associated with resource depletion and low recycling rates, which has promoted the development of circular material systems. Herein, a novel concept is introduced, where a polymer is used as a source of a fertilizer. To demonstrate the viability of this concept, the chemical recycling of poly(isosorbide carbonate) (PIC) is presented as a model for the next generation of plastic-recycling systems. PIC, a bio-based polymer known for its excellent physical properties, undergoes a degradation reaction with aqueous ammonia. The utility of isosorbide and urea obtained from the degradation of PIC as fertilizers was demonstrated via plant-growth experiments. The generation of fertilizers via polymer degradation in the present study is expected to lead not only to innovative chemical recycling systems to address the environmental problems associated with polymer materials, but also to provide solutions to the food-production problems associated with the growth of the global population.
Maleimidophenyl Isocyanate Derivatives as Post-polymerization Modification Agents
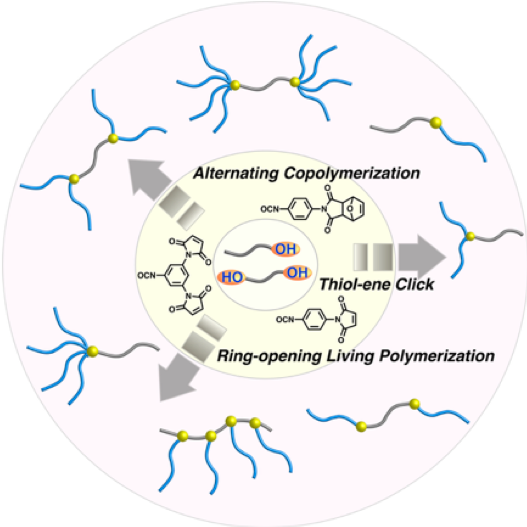
The maleimide structure is highly reactive, exemplified by thiol-ene click reactions with thiols and Diels–Alder reactions with furans. Although post-polymerization modifications and macromolecular conjugations involving maleimide units have been widely studied, mostly due to their selectivity and high reactivity, little has been reported on the one-pot introduction of maleimides in polymer chains. Herein, we report p-maleimidophenyl isocyanate (PMPI) and its derivatives as modification agents to introduce maleimide moieties by reaction with hydroxy groups into polymer chains. The high reactivity of the resulting modification agents and of the corresponding maleimide structures once inserted in the polymer chains was examined by studying their reaction kinetics. Furthermore, these modification agents were successfully applied to the synthesis of macromonomers for graft polymerization and various block copolymers, with e.g. AB-type, star-shaped, and H-shaped architectures.
Characterization of Phenylmaleimide-terminated Poly(ethylene glycol)s and Their Application to a Tetra-arm Poly(ethylene glycol) Gel

The synthesis and characterization of a tetra- PEG gel using a tetra-arm polymer with N-phenylmaleimide moieties at the polymer ends (tetra-N-aryl MA PEG) as a scaffold, were reported. Tetra-N-aryl MA PEG can be obtained via a simple maleimidation using the modification agent p-maleimidophenyl isocyanate (PMPI), which directly transforms the hydroxy groups at the polymer ends into reactive N-aryl maleimide groups in a one-pot reaction. The thus obtained tetra-N-aryl MA PEG was fully characterized using high-performance liquid chromatography (HPLC), matrix-assisted laser desorption ionization time of flight mass spectrometry, and proton nuclear magnetic resonance spectroscopy. HPLC analysis not only demonstrated the high purity of tetra-N-aryl MA PEG and the full conversion of the hydroxy groups, but also provided an effective characterization method for N-aryl maleimide-based PEG using a simple protocol, which enables us quantitative analysis of functionalized polymers with different N-aryl maleimide numbers. Furthermore, we fabricated a TetraPEG gel via Michael addition of the obtained tetra-N-aryl MA and thiol-terminated TetraPEGs.
A Strategy toward Cyclic Topologies Based on the Dynamic Behavior
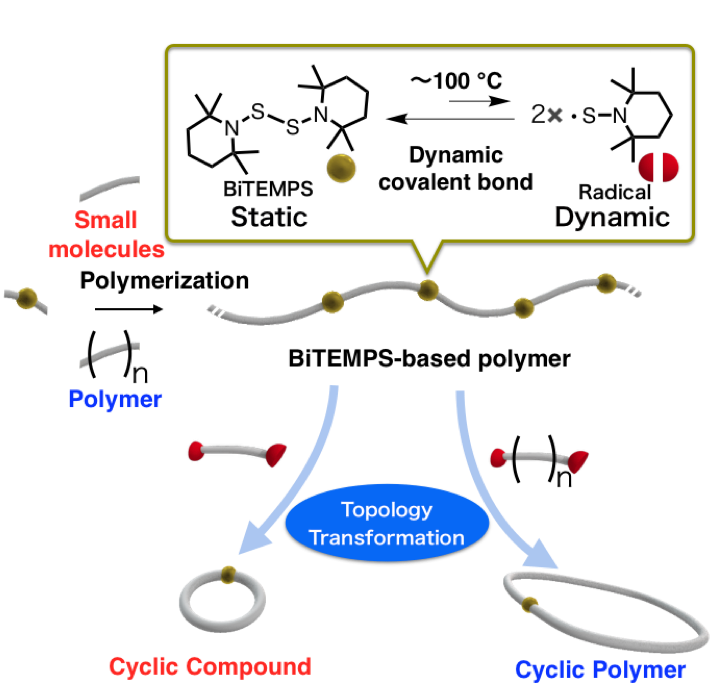
A simple and efficient method to generate macrocyclic structures has been developed based on the dynamic behavior of the linker bis(2,2,6,6-tetramethylpiperidin-1-yl)disulfide (BiTEMPS). The prime linear structure was transformed into a macrocycle using the following sequence: (i) thiol-ene reaction with a BiTEMPS derivative to afford the linear precursor, followed by (ii) an entropy-driven transformation induced by diluting and heating. The radicals generated from BiTEMPS upon heating are highly tolerant toward a variety of chemical species, including oxygen and olefins, while they exhibit high reactivity toward exchange reactions, which can be applied to the topology transformation of various skeletons. The specific advantages of the present method, i.e., its procedural simplicity and substrate versatility, are demonstrated by the gram-scale synthesis of cyclic compounds with low and relatively high molecular weight.
Dynamic Behavior of Cyclic Monomer with Bis(hindered amino)disulfide

Macrocycles (MCM) that contain BiTEMPS units can be obtained on the gram-scale using a relatively small amount of solvent, i.e., it is possible to produce MCMs on a large scale via simple route. The end-functionalized polymers can be obtained by intentionally adding linear structures with functional groups into polymerization system.Cyclic polymers can be also obtained by simply heating MCMs.
Synthetic Strategy for Mechanically Interlocked Cyclic Polymers via the Ring-Expansion Polymerization
Mechanically interlocked polymers that contain rotaxane and catenane structures have attracted much attention on account of their unique properties arising from the restricted mobility of their interlocked structure and their robustness which are comparable to those of covalent bonds. Among these polymers, mechanically interlocked cyclic polymers (MICPs) promise great potential as a novel type of polymers with a large movable area of the interlocked structure. However, synthetic routes to MICPs are not well developed and it is still challenging to create MICPs. The present study has resulted in an effective method for the synthesis of MICPs from the combination of the ring-expansion polymerization (REP) of cyclic disulfide monomers with supramolecular interactions. The results provide guidelines for the successful design of MICPs, i.e., a combination of the dynamic nature of the MMs and supramolecular interac-tions. Given that the present method is highly versatile, it can be expected to be applicable to various molecular skeletons and supramolecular systems.
Topology Transformation of Figure-eight-shaped Polymers Toward Cross-linked Polymers
In this study, various cyclic polymers were synthesized based on the dynamic behavior of the bis(2,2,6,6-tetramethylpiperidin-1-yl)disulfide (BiTEMPS) linkage. Linear and 4-branched polymers were transformed into cyclic topologies, namely, cyclic polymers and figure-eight-shaped polymers. The simplicity and substrate versatility of this procedure are demonstrated via the highly efficient gram-scale synthesis of cyclic and figure-eight-shaped polymers. Moreover, we describe the topology transformation of the obtained cyclic and/or figure-eight-shaped polymers into cross-linked polymers with controlled physical properties by using their cyclic or figure-eight topology and the dynamic nature of the BiTEMPS units in their structures.
Visualization of Slide-ring Effect: A study on Movable Cross-Linking Points Using Mechanochromism

To evaluate the ‘slide-ring’ effect in rotaxane cross-linked network, we incorporated mechanochromophores into static and rotaxane cross-linking points and compared the mechanochromisms exhibited by the obtained polymers. This novel strategy reveals a molecular-level phenomenon with visible color change as well as quantitative electron paramagnetic resonance measurements.
Star-Linear Topological Transformation via Rotaxane Linkage
The development of topologically transformable polymers in response to a specific stimulus is particularly attractive to control polymer properties and to construct novel stimuli-responsive materials. However, most topological polymers reported so far are not transformable, because they are constructed by covalent bonds: there are a few reports on polymer topological reconstruction using weak interactions. We have discussed the construction and function of dynamic macromolecular systems having mechanically linked polymer chains which can undertake topological transformations to induce structure and property changes. The structure definite polyrotaxane, macromolecular [2]rotaxane (M2R) comprising one axle polymer chain and one wheel component, played a crucial role in the fabrication of rotaxane-linked polymers and topologically-transformable polymers. In this study, topology-transformable block copolymers that are connected via rotaxane linkages are introduced. We will present systems in which the topology transformation of block copolymers changes their 1) microphase-separated structures and 2) macroscopic mechanical properties. The combination of a rotaxane structure at the junction point and block copolymers that spontaneously form microphase-separated structures in the bulk provides access to systems that cannot be attained using conventional covalent bonds.
Cyclic-Linear Topological Transformation via Rotaxane Linkage
Cyclic polymers have attracted much scientific and industrial attention, owing to the unique properties that stem from structural characteristics of no chain end. But the synthesis of cyclic polymers has typically suffered from low yields and lack of purity, largely due to the difficulties associated with purification and the selectivity of macrocyclization. Herein, simple and effective synthetic route to cyclic polymers has been developed based on the following sequence: i) selective cyclization of two self-complementary sec-ammonium-containing crown ether monomers to afford [c2] daisy chain bifunctional initiators, ii) living polymerization to afford the corresponding linear polymers, and iii) a topology transformation of these linear polymers to furnish cyclic polymers. The key step in this sequence is the quantitative cyclization via self-assembly of two crown ether molecules with hydroxyl and sec-ammonium moieties. After the living polymerization, the linear polymers release the daisy-chain assembly to generate a cyclic topology. The specific advantages of the present synthetic protocol, i.e., procedural simplicity and concentration independence, are demonstrated by a gram-scale synthesis. A reversible linear-cyclic topological transformation of a poly(ε-caprolactone) facilitated by a long-range rotaxane switch is also reported. A linear polymer, whose topology is fixed by a protonated tert-amine/crown ether interaction at the center of the polymer chain, was reversibly transformed into a cyclic polymer fixed by urethane linkage/crown ether interaction at the polymer end upon neutralization/acidification of the tert-amine moiety.
Rotaxane-cross-linked polymers
Rotaxane-cross-linked polymers (RCPs) have attracted great attention both from a fundamental and a practical perspective due to the extraordinary functionality based on the mechanically linked components with high mobility. We have studied the synthesis and potential applications of crown-ether-containing vinylic rotaxane cross-linkers (RCs) for the synthesis of RCPs by radical polymerization. RCs endow polymers with high toughness, whereby an increasing mobile distance of the RC components improves the mechanical properties of the RCPs and induces unique properties such as high swellability, extensibility, and desirable stress-relaxing ability. As new methods for the toughening of cross-linked polymers have been developed in response to the increasing interest in RCPs, the genuine significance of the component mobility of RCs to endow RCPs with toughness should be clarified in order to firmly establish this new concept as a molecular design guideline.